0導讀:先建立世界觀,再學軟體
這份地圖是給誰的、怎麼讀
很多人一進 Machine Learning Potential(機器學習勢能,常簡稱 MLP 或 MLIP)就直接被丟去裝 NequIP、抄 MACE 的指令、改 input 檔——結果指令會跑了,卻不知道自己在做什麼、為什麼要這樣做。
這份地圖刻意不從軟體教起。它先用七張圖,帶你看懂一條主線:DFT 算得準但太慢 → MLP 學會 DFT 的「結構→能量」對應,又快上千倍 → 用 MLP 跑大尺度 MD 去看真實材料怎麼演化。把這條主線想通了,之後再學任何一個套件,都只是換工具,世界觀是同一套。
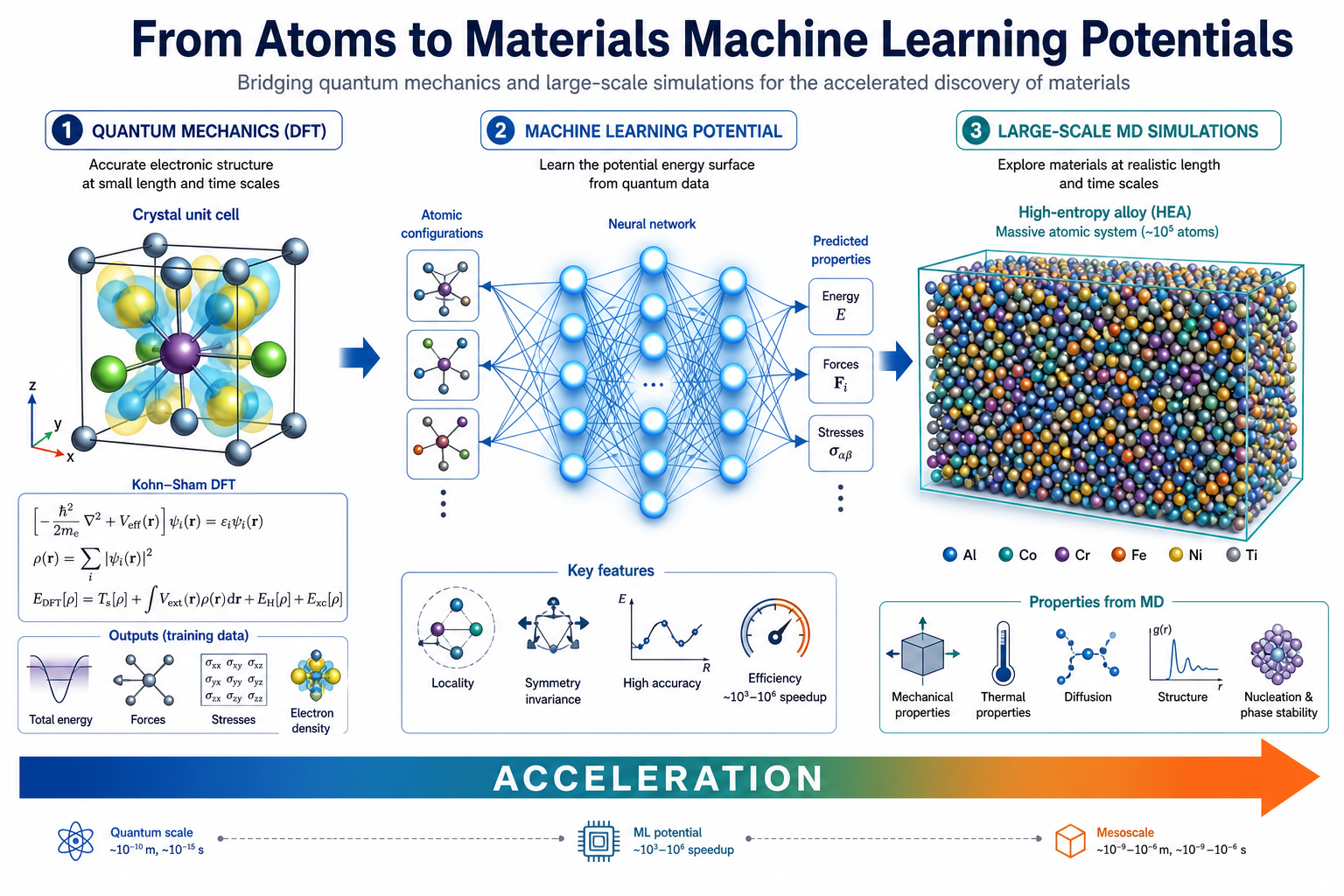
1DFT 為什麼不夠快?
一句話:DFT 每走一步都要重新「解電子」
DFT(密度泛函理論)是計算材料的黃金標準,它直接從量子力學算出每個原子受的力、整個系統的能量,準。但它的代價是:每一個 MD 時間步、每一個結構,都要重新求解一次電子結構。計算量大約隨原子數的三次方(O(N³))成長。
結果就是:用 DFT 跑分子動力學,你大概只能負擔幾百個原子、上千步、跑好幾天。但真實材料的有趣現象——擴散、相變、表面重構、高熵合金的元素偏析——往往需要上萬到十萬個原子、跑上百萬步才看得到。這就是 DFT 的天花板。
MLP 的價值就在這個落差裡:它不取代 DFT 的準確度,而是繞過 DFT 的速度瓶頸,讓你用接近 DFT 的精度,跑到原本只有經典力場才負擔得起的尺度。
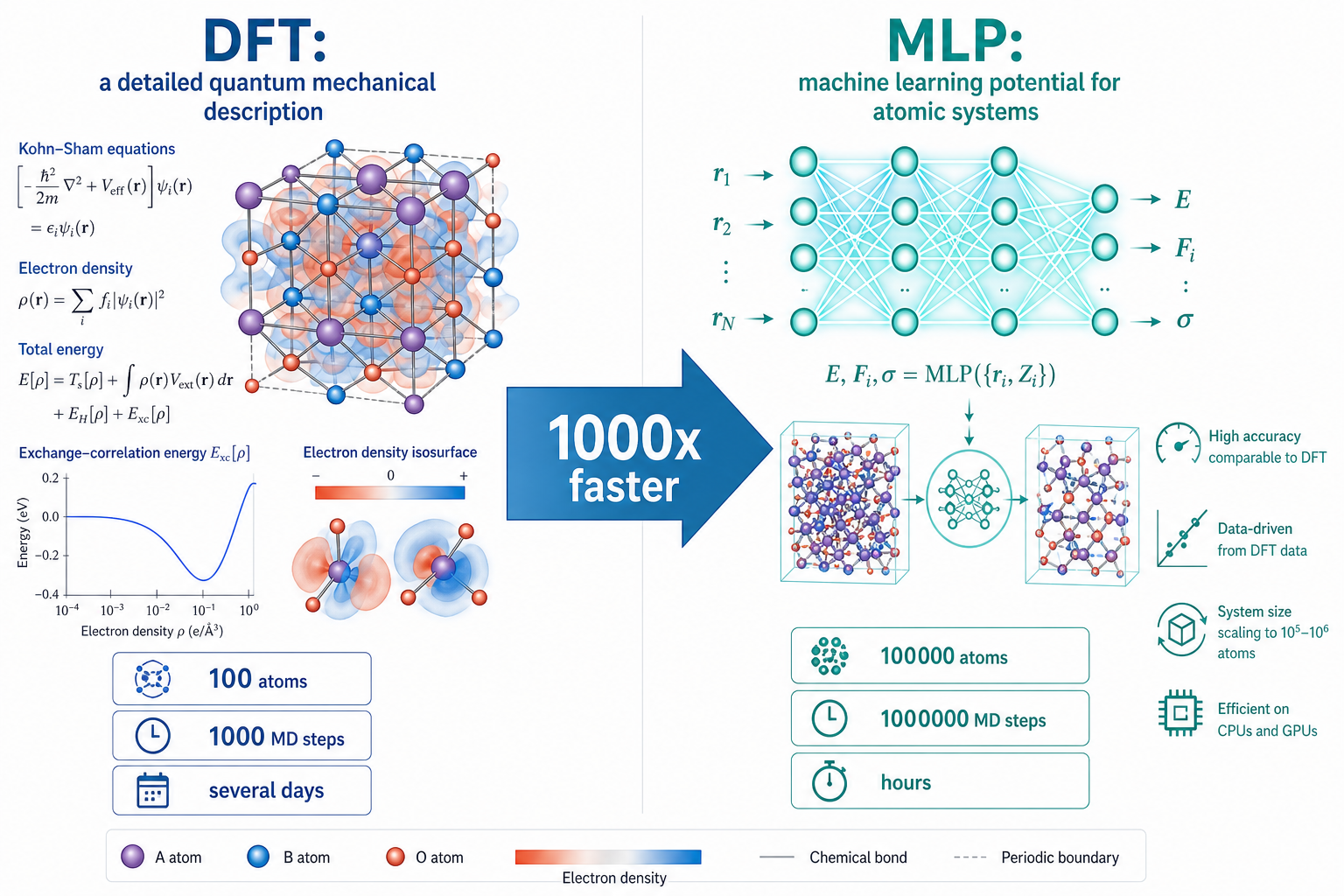
這張圖的生成提示詞(gpt-image-2, medium)
A scientific comparison infographic split into two vertical halves. Left titled DFT: a detailed quantum mechanical crystal structure with equations and electron density clouds, labels 100 atoms, 1000 MD steps, several days. Right titled MLP: a glowing neural network connected to atomic structures, labels 100000 atoms, 1000000 MD steps, hours. A large central arrow labeled 1000x faster. White background, blue teal orange palette, English labels only.
2MLP 到底學到了什麼?
關鍵觀念:它不是在「背材料」,而是在學一個函數
初學者最大的誤解,是以為 MLP「記住了某個材料」。其實不是。MLP 學的是一個數學對應關係:
更精確地說,給定一組原子的位置與種類 R,網路要輸出整個系統的位能 E(R)。一旦能量會算了,力就是能量對座標的負梯度 F = −∂E/∂R,應力則是對應變的微分——這兩個都能由網路自動微分得到。MD 需要的就是「每一步每個原子受的力」,所以一個會算能量與力的 MLP,就足以驅動整場模擬。
因為學的是「局部原子環境 → 能量貢獻」這種可轉移的物理規律,訓練好的 MLP 可以套用到比訓練結構更大的系統上(這叫 size transferability)。這也是它能從百原子訓練、卻拿去跑十萬原子的原因。
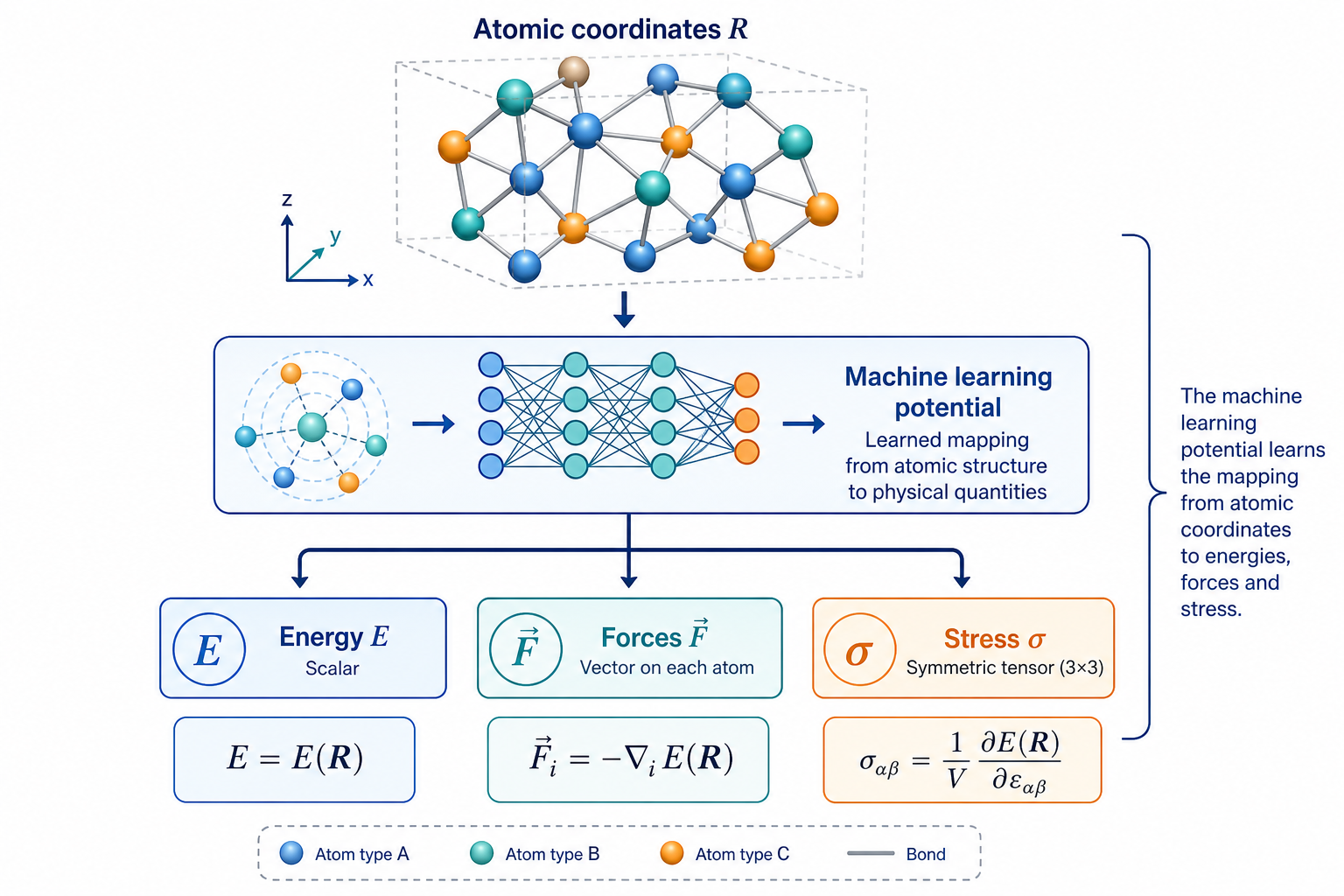
這張圖的生成提示詞(gpt-image-2, medium)
A scientific diagram explaining what a machine learning potential learns. Top: a 3D atomic structure of colorful atoms with bonds, labeled Atomic coordinates R. Arrow down into a neural network block. Outputs three boxes: Energy E, Forces F, Stress sigma. Equations E(R) and F = -gradient of E. White background, blue teal palette, English labels only.
3MLP 是怎麼訓練出來的?
這張流程最重要——資料的源頭永遠是 DFT
MLP 不會憑空變準。它的「標準答案」全部來自 DFT。整個流程是:先用 DFT 算一批結構的能量與力,攢成資料集;拿這批資料去訓練網路,讓它的預測逼近 DFT;再用一批沒看過的結構做驗證,確認誤差夠小;最後才把它放上 LAMMPS 等引擎做大規模 MD。
這裡有兩個新手一定要記住的觀念:
- MLP 的上限就是 DFT 的水準。你用什麼 functional 算資料,MLP 就學到那個 functional 的「世界」。DFT 算錯的,MLP 不會幫你修正。
- 沒看過的結構不要亂信。MLP 擅長內插、不擅長外推。訓練資料沒涵蓋的構型(例如一個從沒出現過的高溫構象),預測可能大錯——這正是第 5 章 Active Learning 要解決的問題。
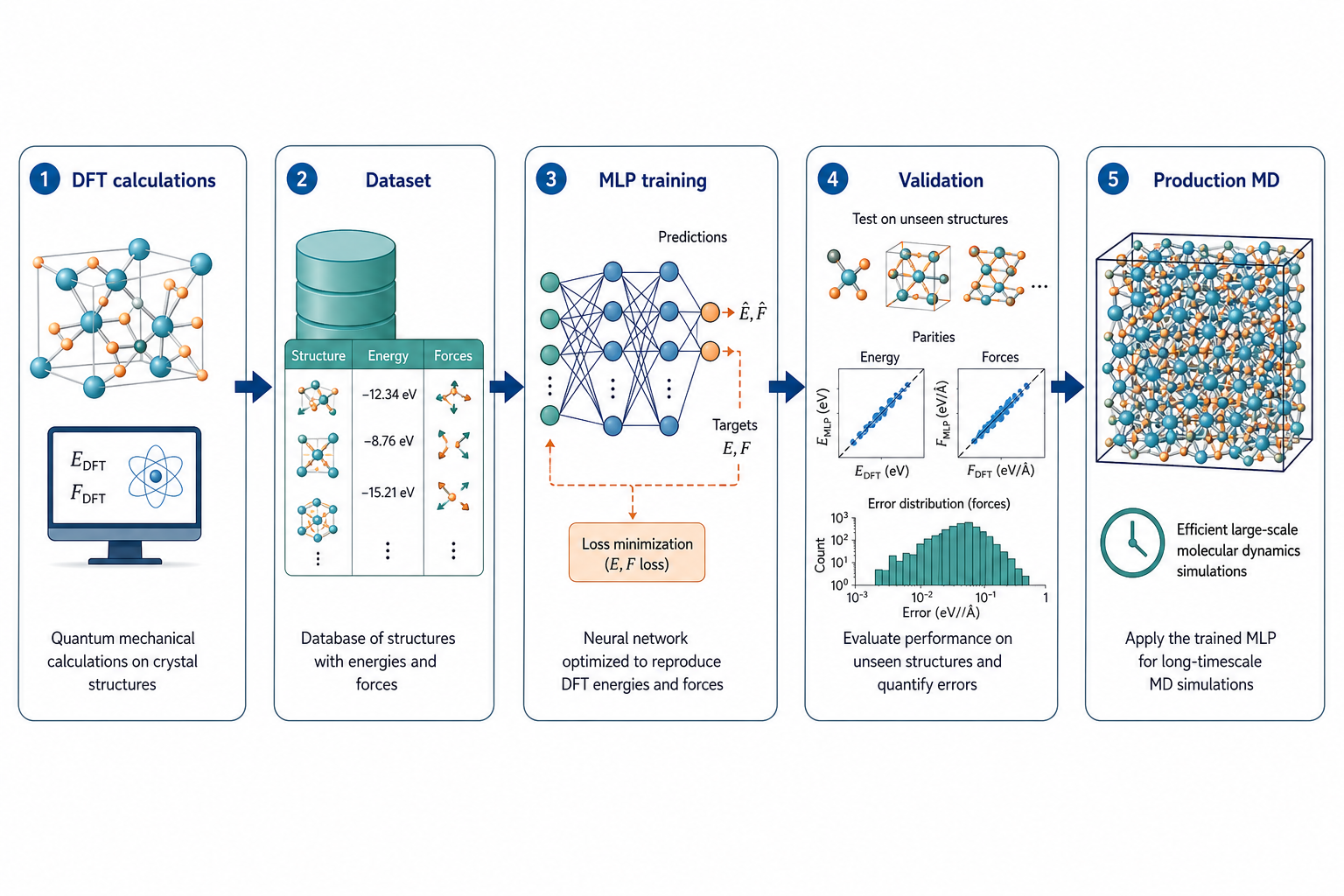
這張圖的生成提示詞(gpt-image-2, medium)
Horizontal left to right workflow of MLP development, five steps with icons and English labels: 1 DFT calculations, 2 Dataset of structures with energies and forces, 3 MLP training neural network, 4 Validation on unseen structures with error plot, 5 Production MD large simulation box. Clean arrows, white background, blue green accents, English labels only.
4DFT → MLP → MD:別把這三個搞混
三者是「真值 / 代理 / 使用者」的關係,不是並列
新手最常混淆的,是把 DFT、MLP、MD 當成三種平行的「軟體選項」。它們其實是一條上下游的鏈:
| 角色 | 它是什麼 | 在鏈中的位置 |
|---|---|---|
| DFT | 真值(Truth)。量子力學算出的「正確答案」 | 提供訓練資料 |
| MLP | 代理模型(Surrogate)。學會模仿 DFT 的快速近似 | 被訓練 → 被 MD 呼叫 |
| MD | 分子動力學。一套讓原子隨時間運動的演算法 | 每一步呼叫 MLP 要力 |
換句話說:MD 是「引擎」,它本身不知道任何物理,每走一步都要問「現在每個原子受多少力?」。傳統上這個問題交給 DFT(準但慢)或經典力場(快但糙)。MLP 的角色,就是當那個又快又接近 DFT 的力計算器,插在 MD 與量子力學之間。
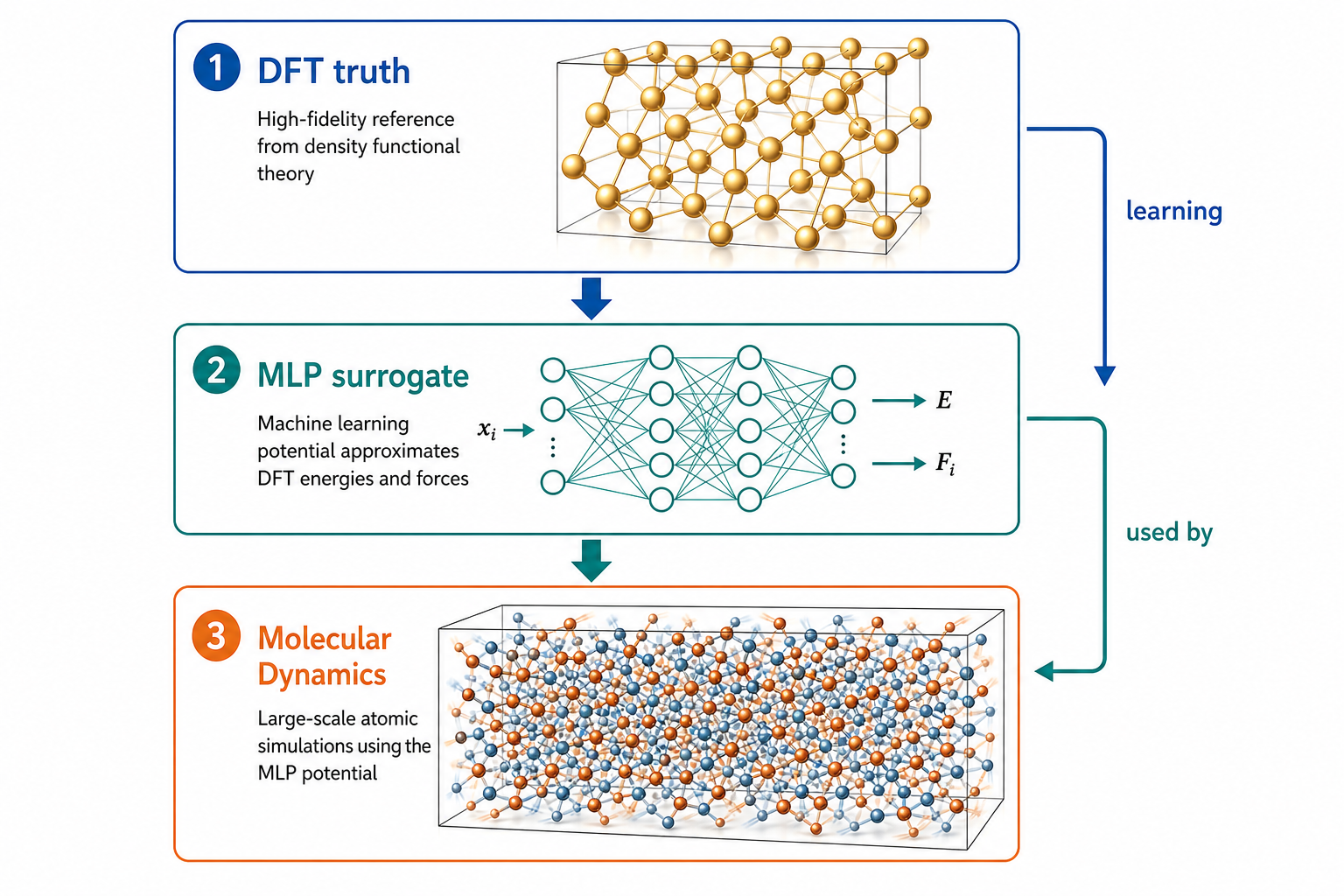
這張圖的生成提示詞(gpt-image-2, medium)
Vertical hierarchy diagram, three levels with downward arrows. Top DFT truth as a golden reference quantum model. Middle MLP surrogate as a neural network. Bottom Molecular Dynamics as thousands of moving atoms. Side arrows labeled learning and used by. White background, English labels only.
5Active Learning:讓 MLP 自己變強
這是這個領域目前最重要的觀念之一
第 3 章說過,MLP 對「沒看過的結構」會失準。Active Learning(主動學習,又叫 on-the-fly learning)就是針對這點的解法:讓 MLP 在跑 MD 的過程中,自己發現哪裡沒把握,然後補課。
運作的循環是這樣:MLP 一邊跑 MD 一邊估計自己的不確定度(常用做法是訓練一組網路看它們彼此分歧多大)。當它撞進一個沒把握的構型,就把那個結構挑出來,回去做 DFT 算出真答案,加進資料集重新訓練。一圈一圈下來,MLP 會越來越涵蓋它真正會遇到的構型空間——變強。
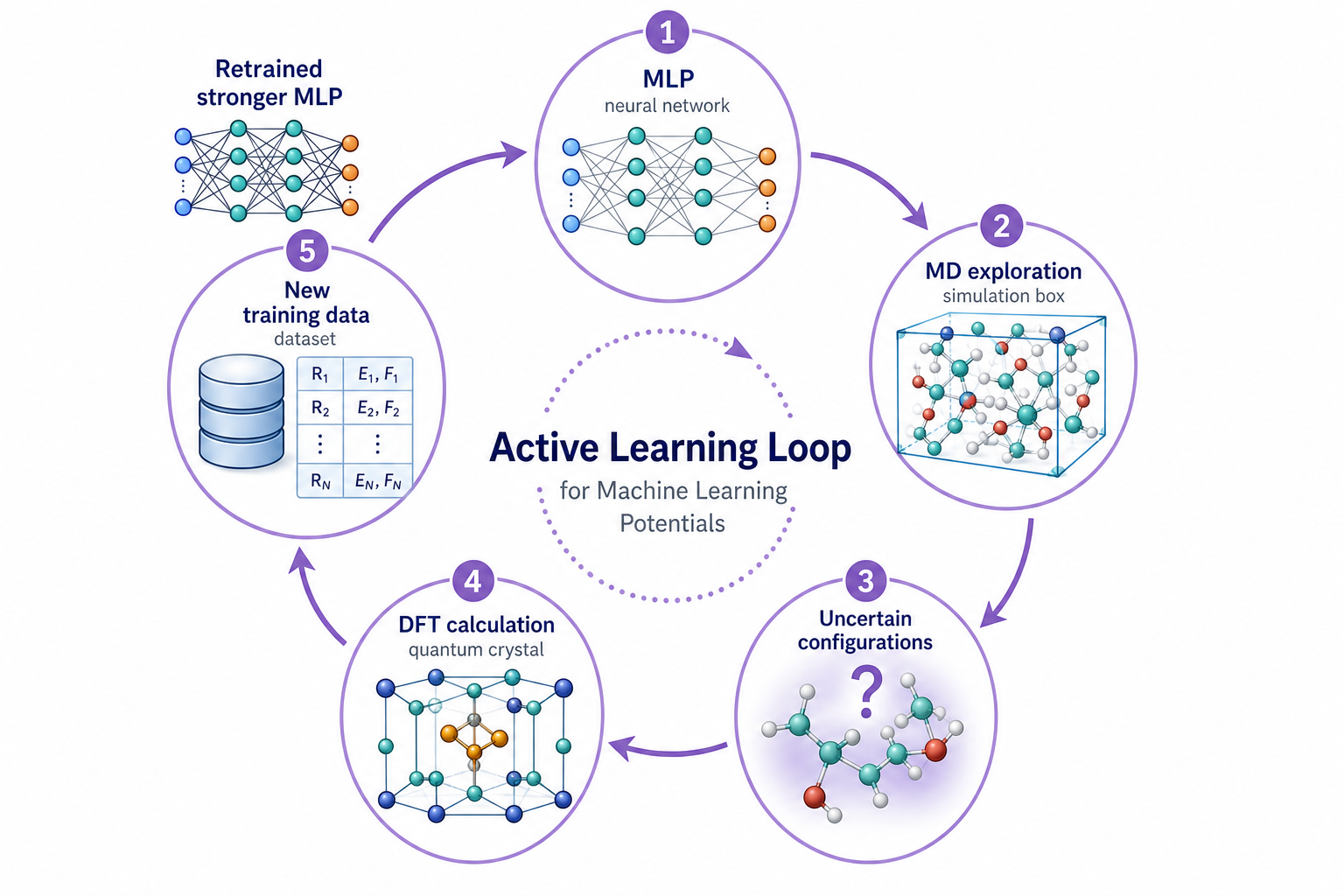
這張圖的生成提示詞(gpt-image-2, medium)
Circular active learning loop for machine learning potentials, purple accent. Five clockwise stages with curved arrows: MLP neural network, MD exploration simulation box, Uncertain configurations structure with question mark and uncertainty halo, DFT calculation quantum crystal, New training data dataset, back to Retrained stronger MLP. White background, English labels only.
6MLP 能做到什麼?四大應用
能跑大、跑久,就能看到 DFT 看不到的現象
一旦你能用接近 DFT 的精度跑十萬原子、上百萬步,很多原本「算不動」的問題就打開了。以下四塊是目前最活躍的方向,其中 HEA(高熵合金)對本實驗室最直接相關。
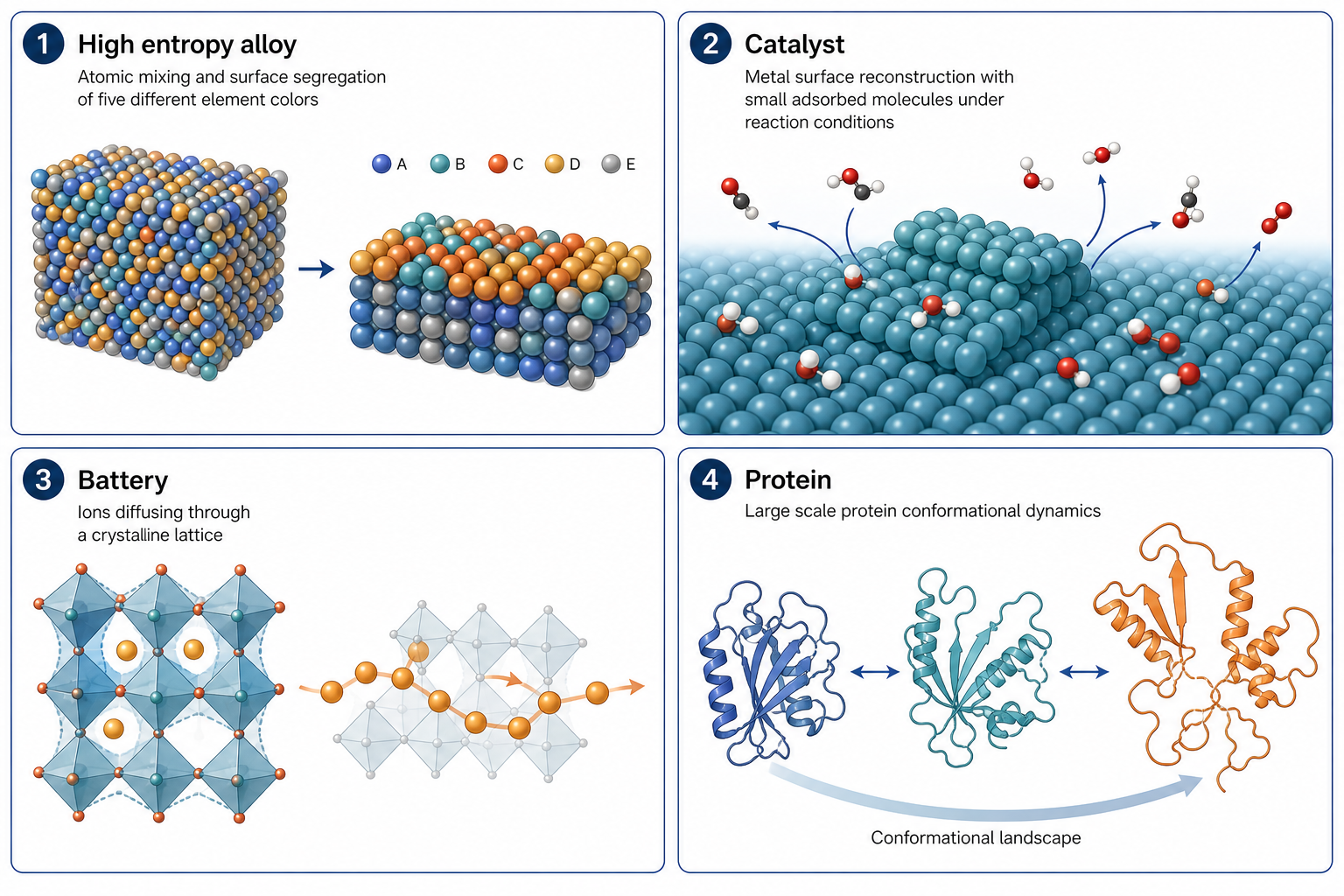
這張圖的生成提示詞(gpt-image-2, medium)
Four panel grid showing MLP applications, each with a short English title. 1 High entropy alloy atomic mixing and surface segregation. 2 Catalyst metal surface reconstruction with adsorbed molecules. 3 Battery ions diffusing through a crystalline lattice. 4 Protein large scale conformational dynamics. Unified style, white background, English labels only.
① HEA 高熵合金 — 本實驗室最相關
五種以上元素隨機混在一起,局部成分千變萬化。MLP 讓你能直接模擬大模型裡的元素交換、退火、短程有序(SRO)、表面偏析——這些都需要大量原子與長時間才看得清楚,正是 DFT 跑不動、經典力場又算不準的甜蜜點。
- 看不同元素在表面/晶界的偏析傾向
- 退火過程中的結構演化與相分離
- 把成分變化連到 d-band center / 吸附能等描述符
② Catalyst 催化 — 動態活性位點
真實催化反應裡,表面不是靜止的。MLP 能模擬反應條件下的表面重構、吸附物造成的局部變形、以及動態活性位點的生成與消失——這些用單一靜態 DFT 結構完全看不到。
③ Battery 電池 — 離子擴散與界面
電池效能取決於離子怎麼在材料裡擴散、在電極/電解質界面發生什麼、以及缺陷如何影響傳輸。這些都是「大尺度+長時間」的統計問題,MLP 驅動的 MD 是目前最實際的工具。
④ Protein 蛋白質 — 大尺度構形變化
生醫端也開始用 MLP(或 ML/MM 混合)去描述需要量子精度的局部反應,搭配大尺度構形變化。這塊較新、仍以經典力場為主流,但方向明確——準確度與尺度兼得。
7一個 HEA 學生的完整工作流程
這張最有價值——它直接對應你進實驗室會做的事
把前面所有觀念串起來,一個做高熵合金的學生,從零到一篇論文的路徑大致是:
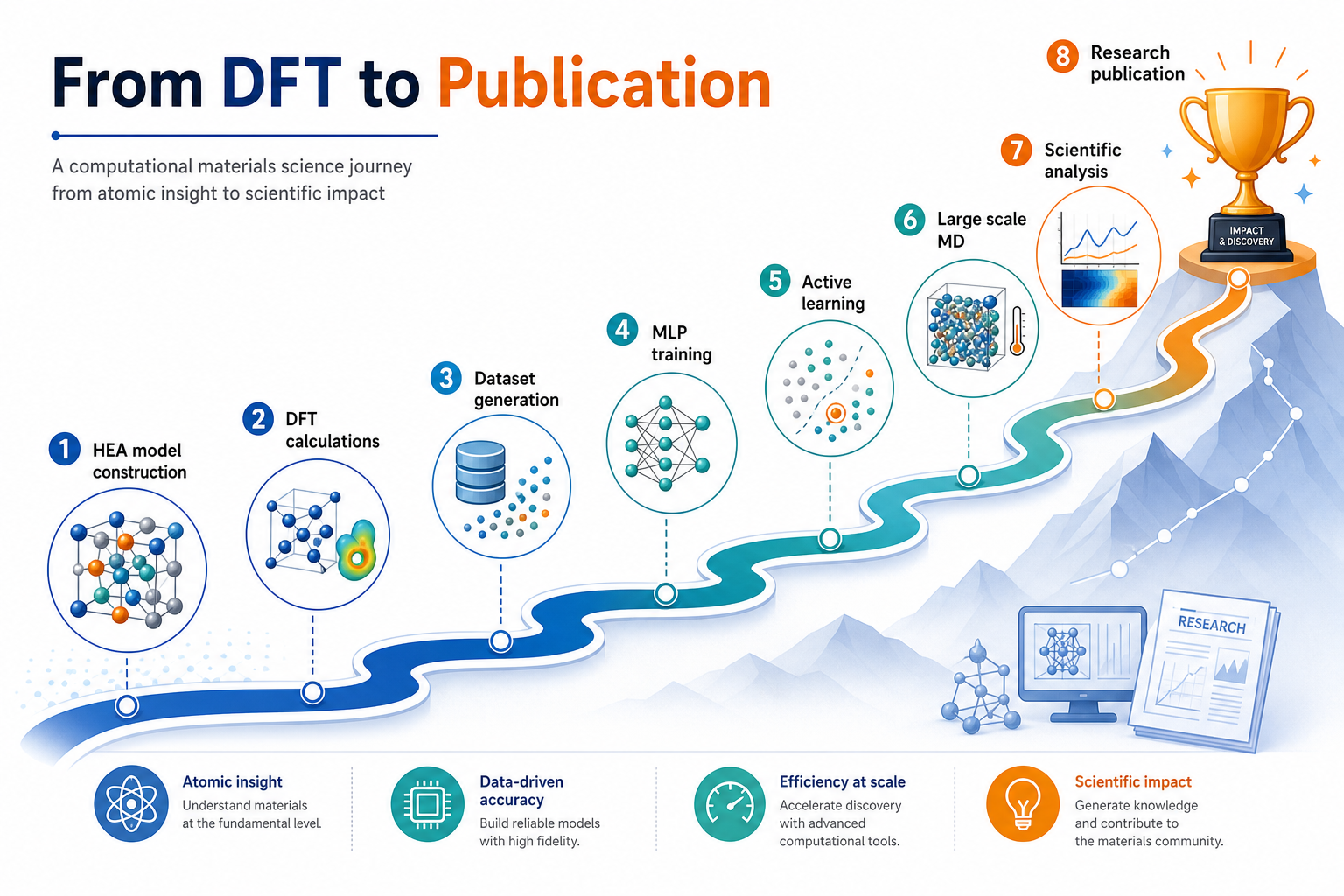
這張圖的生成提示詞(gpt-image-2, medium)
Educational roadmap poster titled From DFT to Publication, the journey of a computational materials science student as a winding upward path with milestone markers and English labels: HEA model construction, DFT calculations, Dataset generation, MLP training, Active learning, Large scale MD, Scientific analysis, Research publication with a trophy. Motivational, clean, white background, English labels only.
★MLP 學習路徑:Level 0 → 5
給本實驗室零基礎學生的進階順序——一階一階往上爬
不要一開始就衝 MACE 指令。照這條階梯走,每一階都先跑得起來、看得懂結果,再上下一階。
什麼是 MD
先搞懂分子動力學在做什麼:原子、力、時間步、溫度、軌跡。不碰 ML,先有「原子會動」的直覺。
LAMMPS 上手
學會用 LAMMPS 跑一個經典力場的 MD:寫 input、設邊界條件、看輸出、用 OVITO 看軌跡。先讓模擬跑得動。
DFT 資料生成
用 VASP 算一批結構的能量與力,理解 functional、收斂、k-point。這是 MLP 的「標準答案」來源。
MACE / NequIP 訓練
拿 Level 2 的資料訓練第一個 MLP,學會看 loss、能量/力誤差,把訓練好的勢能接回 LAMMPS 跑 MD。
Active Learning
建立不確定度估計與「自動補 DFT」的循環,讓 MLP 在你關心的構型空間裡自己變可靠。
HEA 論文
把整條鏈用在真實高熵合金題目上:大尺度 MD、分析偏析/SRO/描述符,產出可發表的結果。
¶名詞速查
看到不認得的詞,回這裡查
✓結語
From Atoms to Materials
把這份地圖讀完,你應該能用一句話回答:「MLP 是什麼?」——它是一個學會模仿 DFT、但快上千倍的力計算器,讓我們用接近量子精度的水準,去跑原本算不動的大尺度、長時間模擬,看見真實材料怎麼演化。
之後不管你用 MACE 還是 NequIP、跑 HEA 還是電池,世界觀都是同一套:DFT 給真值 → MLP 學成代理 → MD 拿去用 → Active Learning 補漏。工具會換,這條主線不變。